
AutoGrow4
AutoGrow4 is an open-source program for semi-automated computer-aided drug discovery. It uses a genetic algorithm to evolve predicted ligands on demand and so is not limited to a virtual library of pre-enumerated compounds. AutoGrow4 is a useful tool for generating entirely novel drug-like molecules and for optimizing preexisting ligands.
BINANA
BINANA (BINding ANAlyzer) analyzes the geometries of predicted ligand poses to identify molecular interactions that contribute to binding. It is useful because accurately characterizing these interactions allows medicinal chemists to assess whether a predicted ligand merits further study. We have also created a BINANA web-browser application so students and chemical-biology researchers can quickly visualize receptor/ligand complexes and their unique binding interactions.
BlendMol
BlendMol is a Blender plugin that can easily import VMD ‘Visualization State’ files. BlendMol empowers scientific researchers and artists by marrying molecular visualization and industry-standard rendering techniques. The plugin works seamlessly with a popular analysis program (VMD). Users can import into Blender the very molecular representations they set up in VMD.
CENsible
CENsible uses deep-learning context explanation networks (CENs) to predict small-molecule binding affinities. Rather than predict a binding affinity directly, it predicts the contributions of pre-calculated terms to the overall affinity, thus providing interpretable output. CENsible insights are useful for subsequent lead optimization.
DeepFrag
DeepFrag is a deep convolutional neural network that guides ligand optimization by extending a ligand with a molecular fragment, such that the resulting extension is also highly complementary to the receptor. The DeepFrag web application is also a useful tool for teaching students about medicinal chemistry and lead optimization.
DeepFrag2
DeepFrag2 is a deep convolutional neural network that guides ligand optimization by extending a ligand with a molecular fragment, such that the resulting extension is also highly complementary to the receptor.
Dimorphite-DL
Dimorphite-DL adds hydrogen atoms to molecular representations, as appropriate
for a user-specified pH range. It is a fast, accurate, accessible, and modular
open-source program for enumerating small-molecule ionization states.
EnOpt
Ensemble Optimizer (EnOpt) is a fast, accessible tool that streamlines ensemble-docking and consensus-score analysis. EnOpt takes as input a matrix of docking scores from an ensemble virtual screen, organized as compounds (rows) X protein conformations (columns). It uses simple, interpretable machine learning to identify most-predictive subensembles and an ensemble composite score.
FPocketWeb
FpocketWeb is a browser app for identifying pockets on protein surfaces where small-molecule ligands (e.g., drugs) might bind. It runs the fpocket executable entirely in a web browser. The pocket-finding calculations occur on the user’s computer rather than a remote server.
Gypsum-DL
Gypsum-DL is a free, open-source program that converts 1D and 2D small-molecule representations (SMILES strings or flat SDF files) into 3D models. It outputs models with alternate ionization, tautomeric, chiral, cis/trans isomeric, and ring-conformational states.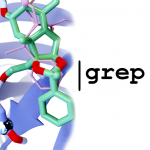
LigGrep
LigGrep is a free, open-source program for identifying docked poses that participate in user-specified receptor/ligand interactions. As input, LigGrep accepts a protein receptor file (PDB, PDBQT), a directory containing many docked-compound files (PDB, PDBQT, SDF), and a list of user-specified filters (JSON). It evaluates each docked pose and outputs the names of the compounds with poses that pass all filters.
MolModa
Molecular docking advances early-stage drug discovery by predicting the geometries and affinities of small-molecule compounds bound to drug-target receptors. Researchers can leverage these predictions to prioritize drug candidates for experimental testing. MolModa provides a secure, accessible environment where users can perform molecular docking entirely in their web browsers.
PCAViz
PCAViz is an open-source Python/JavaScript toolkit for sharing and visualizing MD trajectories via a web browser. It is also a helpful tool for class websites and presentations. To encourage use, an easy-to-install PCAViz-powered WordPress plugin enables ‘plug-and-play’ trajectory visualization.
POVME2
POVME2 (POcket Volume MEasurer 2) scans a molecular-dynamics simulation and extracts druggable protein pockets with unique conformations. It is useful because ligand-binding pockets adopt many conformations in response to atomic forces. Characterizing these pocket geometries helps identify those that are particularly amenable to ligand binding. Computer-aided drug-discovery campaigns that incorporate these conformations often identify novel pharmacologically active molecules.
Prot2Prot
Prot2Prot is a deep-learning model trained to imitate a Blender-rendered image given a much simpler representation (‘sketch’) of a protein surface. Prot2Prot outputs an image that is often indistinguishable from a Blender/BlendMol-based visualization in a fraction of the time, allowing image ‘rendering’ even in a web browser.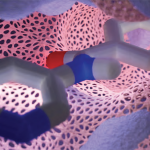
ProteinVR
ProteinVR is a web-based application that allows users to view protein/ligand structures in virtual reality (VR) from their mobile, desktop, or VR-headset-based web browsers. Molecular structures are displayed within 3D environments that give useful biological context and allow users to situate themselves in 3D space. We expect the ProteinVR web application to be useful in both research and classroom settings.
Pyrite
Pyrite is a Blender plugin that imports the atomic motions captured by molecular dynamics simulations. Using Pyrite, these motions can be rendered using Blender’s state-of-the-art computer-graphics algorithms. All 3D protein representations (e.g., surface, ribbon, VDW spheres) are supported. Aside from advancing scientific and collaborative objectives, Pyrite renderings will also appeal to students and non-specialists.
Scoria
Scoria is a Python package for manipulating three dimensional molecular data. Unlike similar packages, Scoria is written in pure Python and so requires no dependencies or installation. One can incorporate the Scoria source code directly into their own programs. But Scoria is not designed to compete with other similar packages. Rather, it complements them. Our package leverages others (e.g., NumPy, SciPy, MDAnalysis), if present, to speed and extend its own functionality.
SubPEx
SubPEx (Sub-Pocket Explorer) is a tool to enhance ensemble (multiple-receptor/relaxed-complex) virtual screening. It uses weighted ensemble path sampling and molecular dynamics simulations to accelerate binding-pocket sampling.
Webina
Webina is a JavaScript/WebAssembly library that runs AutoDock Vina entirely in a web browser. The docking calculations take place on the user’s own computer rather than a remote server. To encourage use, we have incorporated the Webina library into our own Webina web application. The app includes a convenient interface so users can easily setup their docking runs and analyze the results. Aside from its research applications, Webina is an excellent tool for teaching students about computer-aided drug discovery, protein/ligand interactions, and computational structural biology.

